
| โรคจอตามีสารสี (Retinitis pigmentosa) | |
|---|---|
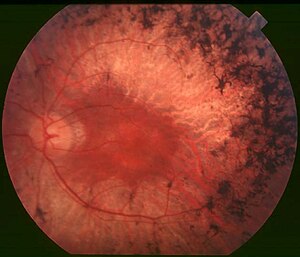 | |
| ด้านหลัง/ก้นตา (Fundus) ของคนไข้โรคอาร์พีระยะกลาง (mid stage) ให้สังเกตสารสี/รอยเปรอะสีดำ (bony spicule) ที่ส่วนรอบ ๆ ตรงกลางที่เกิดกับการเสื่อมของจอตา (retinal atrophy) แม้จุดภาพชัด (macula) จะคงสภาพอยู่แต่ส่วนรอบ ๆ ก็ซีดไป เส้นเลือดในจอตาก็ลีบลงด้วย | |
| สาขาวิชา | จักษุวิทยา |
| อาการ | เห็นไม่ดีต่อกลางคืน เห็นรอบนอก (peripheral vision) ได้แย่ลง |
| การตั้งต้น | ตั้งแต่เด็ก |
| สาเหตุ | กรรมพันธุ์ |
| วิธีวินิจฉัย | การตรวจตา |
| การรักษา | อุปกรณ์ช่วยคนที่เห็นได้น้อย ไฟส่องที่พกพาได้ สุนัขนำทาง |
| ยา | Vitamin A palmitate |
| ความชุก | 1 คนใน 4,000 คน |
โรคจอตามีสารสี หรือ โรคอาร์พี (อังกฤษ: Retinitis pigmentosa ตัวย่อ RP) เป็นความผิดปกติทางพันธุกรรมทางตาที่ทำให้เสียสายตา อาการรวมทั้งตาฟางกลางคืน (nyctalopia) และการเสียการเห็นรอบนอก (peripheral vision) คือเห็นข้าง ๆ หรือรอบ ๆ ได้น้อยลง อาการปกติจะค่อย ๆ ปรากฏ เมื่อเห็นรอบนอกได้แย่ลง บางคนอาจลานสายตาแคบลงเหมือนมองผ่านอุโมงค์ (tunnel vision) แต่ตาบอดสนิทจะไม่สามัญ
โรคปกติจะสืบทอดมาจากพ่อแม่ โดยมีการกลายพันธุ์ของยีนหนึ่ง ๆ ในบรรดา 50 ยีนที่ได้ระบุแล้วเป็นอย่างน้อย วิถีการดำเนินของโรครวมการค่อย ๆ เสียเซลล์รับแสงรูปแท่งที่ด้านหลังของตา แล้วปกติจะตามด้วยการเสียเซลล์รูปกรวยต่อมาการวินิจฉัยจะทำเมื่อตรวจจอตาแล้วพบสารสี/รอยเปรอะดำ วิธีการตรวจยืนยันอื่น ๆ รวมทั้ง electroretinography, การตรวจลานสายตา และการตรวจยีน
ปัจจุบันยังไม่มีวิธีรักษาโรคนี้ วิธีช่วยบรรเทาปัญหาในชีวิตรวมทั้งอุปกรณ์ช่วยการมองเห็น (เช่น แว่นขยาย) ไฟส่องที่พกพาได้ (เช่น ไฟฉาย) และสุนัขนำทาง อาหารเสริมคือ vitamin A palmitate อาจช่วยให้สายตาแย่ช้าลง ตาเทียม (visual prosthesis) อาจเป็นทางเลือกอย่างหนึ่งสำหรับคนที่มีโรครุนแรงมาก ประเมินว่าคน 1 คนใน 4,000 คนมีโรค โรคจะเริ่มตั้งแต่วัยเด็กแต่บางคนอาจไม่ได้รับผลกระทบจนกระทั่งเป็นผู้ใหญ่
ผลของโรคเห็นได้ง่ายถ้าเปรียบเทียบกับทีวีหรือจอคอมพิวเตอร์ คือ แสงจากพิกเซลที่สร้างภาพบนจอเหมือนกับเซลล์รับแสงเป็นล้าน ๆ ตัวในจอตา ยิ่งมีพิกเซลน้อยลงเท่าไร ภาพที่เห็นก็ชัดน้อยลงเท่านั้น มีเซลล์รับแสงจำนวนน้อยกว่า 10% ที่สามารถรับภาพสี โดยเป็นแสงสว่างดังที่มีในช่วงกลางวัน เซลล์เหล่านี้อยู่ตรงกลางของจอตา เซลล์รับแสงกว่า 90% ที่เหลือสามารถรับแสงสลัวเป็นภาพขาวดำ ซึ่งใช้ในที่สลัวและในตอนกลางคืน เป็นเซลล์ซึ่งอยู่รอบ ๆ จอตา โรคทำลายเซลล์รับแสงจากนอกเข้ามาส่วนตรงกลาง หรือจากส่วนตรงกลางออกไปส่วนนอก หรือทำลายเป็นย่อม ๆ ที่ทำให้เซลล์ส่วนตรงนั้นมีประสิทธิภาพตรวจจับแสงได้น้อยลง ความเสื่อมจากโรคนี้จะลุกลามโดยยังไม่มีวิธีรักษา
อาการ

อาร์พีเป็นโรคที่เกิดอาการเพราะเสียเซลล์รับแสงอันบุอยู่ที่ด้านหลังของดวงตาและทำหน้าที่คล้ายกับฟิลม์ในกล้องถ่ายรูป โดยปกติแล้ว เซลล์รูปแท่งซึ่งมีหน้าที่เกี่ยวกับการเห็นเวลากลางคืน มักจะเสียหายก่อน ซึ่งอธิบายว่า ทำไมความมีตาบอดแสง (nyctalopia) จึงเป็นอาการที่ปรากฏขึ้นก่อน การเห็นเวลากลางวันซึ่งอำนวยโดยเซลล์รูปกรวยจะคงสภาพจนถึงระยะหลัง ๆ ของโรค ความเปรอะเปื้อนของ retinal pigment epithelium ด้วยสีดำ ๆ (เหมือนกระดูกและเข็ม) เป็นอาการทั่ว ๆ ไปของโรค ลักษณะที่เห็นได้ในตาอย่างอื่นคือจานประสาทตา (optic disc) ปรากฏเป็นสีเหลืองซีด และเส้นเลือดเกิดตีบแคบ
อาการเสื่อมของจอตาเบื้องต้นก็คือตาฟางกลางคืน (nyctalopia) และการเสียการเห็นที่ลานสายตารอบนอกถัดเข้ามา (mid-peripheral visual field) เซลล์รับแสงรูปแท่ง ซึ่งทำหน้าที่ในแสงสลัวและมีอยู่โดยมากที่รอบ ๆ จอตา เป็นส่วนที่ได้รับผลก่อนเพื่อนในโรคแบบที่ไม่เกิดร่วมกับความผิดปกติอื่น ๆ (non-syndromic) ตาจะแย่ลงค่อนข้างเร็วโดยขยายไปถึงลานสายตารอบนอกสุด แล้วในที่สุดก็จะเข้ามาถึงส่วนตรงกลางทำให้เห็นเหมือนผ่านอุโมงค์มากขึ้น ความคมชัดและการเห็นเป็นสีอาจแย่ลงเพราะความผิดปกติของเซลล์รูปกรวย ซึ่งมีหน้าที่เกี่ยวกับการเห็นเป็นสี ความคมชัด และการเห็นส่วนตรงกลาง โรคอาจจะดำเนินไปอย่างเท่า ๆ กันทั้งสองข้าง คือตาซ้ายและขวาจะเกิดอาการคล้ายกัน นอกจากผลโดยตรงที่เกิดจากความเสื่อมของเซลล์รูปแท่งและต่อมาเซลล์รูปกรวย ก็ยังมีผลโดยอ้อมอื่น ๆ ซึ่งปรากฏเป็นอาการของโรค ปรากฏการณ์เช่น อาการกลัวแสง (photophobia) คือมองเห็นแสงธรรมดาเป็นแสงจ้ามาก และ photopsia คือมองเห็นแสงกระพริบหรือแสงวาบ (เช่นเหมือนฟ้าแลบ) ภายในลานสายตา บ่อยครั้งจะเกิดในระยะหลัง ๆ ของโรค มีอาการ 3 อย่างที่ก้นตา (fundus) ซึ่งเป็นลักษณะเฉพาะของโรคที่เรียกว่า ophthalamic triad คือ
- ชั้น retinal pigment epithelium (RPE) ของจอตาจะปรากฏเป็นรอยเปรอะดำเพราะเกิด bony spicule
- จานประสาทตา (optic disc) ปรากฏเป็นสีเหลืองซีดคล้าย ๆ ขี้ผึ้ง
- หลอดเลือดในจอตาจะลีบลงลง
โรคที่ไม่เกิดร่วมกับความผิดปกติอื่น ๆ (Nonsyndromic RP) ปกติจะมีอาการต่าง ๆ ดังต่อไปนี้
- ตาฟางกลางคืน (nyctalopia)
- ลานสายตาแคบลงเหมือนมองผ่านอุโมงค์/การไม่เห็นรอบนอก (tunnel vision)
- Latticework vision
- photopsia คือมองเห็นแสงกระพริบหรือแสงวาบ (เช่น เหมือนฟ้าแลบ) ภายในลานสายตา
- อาการกลัวแสง (photophobia) คือมองเห็นแสงธรรมดาเป็นแสงจ้ามาก
- การเกิดรอยเปรอะดำคือ bony spicule (การสะสมสารสีเป็นรูปโครงมีปลายเหมือนเข็ม) ที่ก้นตา
- การปรับตัวจากที่มืดเป็นที่สว่าง และนัยตรงกันข้ามได้ช้า
- การเห็นมัว
- แยกสีได้ไม่ดี
- การเสียการเห็นตรงกลาง
- ในที่สุดจะจัดว่าบอด
เหตุ
โรคอาจจะเป็น
- Nonsyndromic คือเกิดโดยลำพังโดยไม่พบอาการอื่น ๆ
- Syndromic คือพบกับความผิดปกติของการรับความรู้สึกทางประสาทอื่น ๆ, กับความผิดปกติทางพัฒนาการ หรือกับอาการที่ซับซ้อนอื่น ๆ
- เกิดเป็นอาการทุติยภูมิ (secondary) ของโรคทั่วร่างกายอื่น ๆ
อนึ่ง
- อาร์พีเมื่อเกิดกับหูหนวก (ไม่ว่าจะแต่กำเนิดหรือค่อย ๆ เป็น) เรียกว่า Usher syndrome
- กลุ่มอาการอัลพอร์ตที่สัมพันธ์กับอาร์พี เป็นโรคความผิดปกติที่ basement membrane ของ glomerulus ซึ่งทำให้มีอาการไตเสื่อม โรคจะสืบทอดแบบ X-linked dominant
- อาร์พีเมื่อเกิดกับอัมพาตกล้ามเนื้อตา (ophthalmoplegia), การกลืนลำบาก (dysphagia), ภาวะกล้ามเนื้อเสียสหการ (ataxia) และความผิดปกติการนำไฟฟ้าที่หัวใจ จะพบในคนไข้ Kearns-Sayre syndrome หรือ Ragged Red Fiber Myopathy โดยเป็นความผิดปกติทางดีเอ็นเอของไมโทคอนเดรีย
- อาร์พีเมื่อเกิดกับปัญญาอ่อน, โรคเส้นประสาทนอกประสาทกลาง (peripheral neuropathy), acanthotic (spiked) RBCs คือเม็ดเลือดแดงที่มีเยื่อหุ้มเซลล์ยื่นออกเป็นหนาม, steatorrhea คือภาวะไขมันเกินในอุจจาระ และการขาดลิโพโปรตีนหนาแน่นต่ำมาก (VLDL) จะพบในคนไข้ abetalipoproteinemia ซึ่งเป็นภาวะขัดขวางการดูดซึมไขมันและวิตามินที่ละลายในไขมันจากอาหาร
- ในทางคลินิก อาร์พีเกิดกับความผิดปกติของยีนที่มีน้อยหลายอย่าง รวมทั้งโรคกล้ามเนื้อเจริญผิดเพี้ยน (muscular dystrophy) และ chronic granulomatous disease โดยเป็นส่วนของ McLeod syndrome นี่เป็นฟิโนไทป์แบบ X-linked recessive ปรากฏเป็นการขาดโปรตีน XK ที่ผิวเซลล์อย่างสิ้นเชิง และดังนั้น จึงมีการเแสดงออกของแอนติเจนแบบ Kell น้อยมากที่เม็ดเลือดแดง เมื่อต้องถ่ายเลือด คนไข้เหล่านี้จึงมีเลือดที่จัดว่าไม่เข้ากับผู้บริจาคเลือดปกติ และผู้บริจาคเลือด K0/K0 โดยสิ้นเชิง
- โรคที่สัมพันธ์กับการทำงานน้อยผิดปกติของอัณฑะหรือรังไข่ (hypogonadism) ทำให้พัฒนาการอย่างล่าช้า และสืบทอดทางพันธุกรรมแบบทายกรรมลักษณะด้อย (autosomal recessive) จะพบในคนไข้โรค Bardet-Biedl syndrome เป็นความผิดปกติทางพันธุกรรมเกี่ยวกับซีเลียของเซลล์และมีผลต่อระบบต่าง ๆ หลายอย่างในร่างกาย
ภาวะอื่น ๆ ที่อาจมีอาการอาร์พีรวมทั้งซิฟิลิสระบบประสาท (neurosyphilis), โรคติดเชื้อท็อกโซพลาสมา (toxoplasmosis) และโรคติดเชื้ออะแคนตามีบา (Refsum's disease)
พันธุศาสตร์
โรคอาร์พีเป็นโรคจอตาเสื่อมอันสืบทอดทางพันธุกรรมซึ่งสามัญที่สุด และปรากฏเป็นความสูญเสียเซลล์รับแสงที่ลุกลาม และอาจจะทำให้ตาบอดได้โดยที่สุด
มียีนหลายยีนที่เมื่อกลายพันธุ์ อาจเป็นเหตุของโรค รูปแบบการสืบทอดทางพันธุกรรมของโรครวมทั้งทายกรรมลักษณะเด่น (autosomal dominant), ทายกรรมลักษณะด้อย (autosomal recessive), X-linked, สืบจากแม่ (โดยไมโทคอนเดรีย) โดยจะขึ้นอยู่กับการกลายพันธุ์ของยีนอาร์พีที่มีในพ่อแม่
ในปี 1989 นักวิจัยได้พบการกลายพันธุ์ของยีนโรด็อปซิน (rhodopsin) ซึ่งเป็นสารสี (pigment) ที่จำเป็นในการถ่ายโอนแสงที่รับทางตาให้เป็นกระแสประสาท (visual transduction cascade) เพื่อให้เห็นในที่สลัว ยีนนี้เข้ารหัสโปรตีนหลักชนิดหนึ่งที่ส่วนนอก (outer segment) ของเซลล์รับแสง การกลายพันธุ์ของยีนที่สามัญที่สุดเป็น missense mutation หรือการพับตัวผิดปกติ (misfolding) ของโปรตีนโรด็อปซิน และปกติจะสืบทอดทางพันธุกรรมในรูปแบบทายกรรมลักษณะเด่น (autosomal dominant) ตั้งแต่ได้ค้นพบยีนนี้ (มีสัญลักษณ์ RHO) ก็ได้ค้นพบการกลายพันธุ์ของยีนถึง 100 รูปแบบ ซึ่งเป็นเหตุของจอตาเสื่อม (retinal degeneration) รูปแบบต่าง ๆ ถึง 15% และเป็นเหตุของอาร์พีที่สืบทอดแบบทายกรรมลักษณะเด่นถึง 25%
ในปี 1990 มีการค้นพบการกลายพันธุ์ Pro23His ของยีนอ็อปซิน (opsin) เบสเดียวที่ intradiscal domain ซึ่งสัมพันธ์กับโรคอาร์พี และจนกระทั่งถึงทุกวันนี้ได้มีการระบุการกลายพันธุ์ของยีนกว่า 150 อย่าง การกลายพันธุ์เหล่านี้พบทั่วยีนอ็อปซินโดยกระจายไปตามโดเมน 3 โดเมนของโปรตีน คือ intradiscal domain, transmembrane domain และ cytoplasmic domain เหตุทางชีวเคมีของโรคในกรณีที่ยีนโรด็อปซินกลายพันธุ์ก็คือการพับตัวผิดปกติของโปรตีน (protein misfolding) และการขัดการทำงานของ molecular chaperones ซึ่งช่วยในการพับโปรตีน
งานต้นคริสต์ทศวรรษ 2000 ต่อ ๆ มาพบว่า การกลายพันธุ์ที่โคดอน (codon) 23 ในยีนโรด็อปซิน ซึ่งเป็นการเปลี่ยนโปรลีน (proline) เป็นฮิสตาดีน (histidine) เป็นการกลายพันธุ์ของยีนโรด็อปซินซึ่งเกิดบ่อยที่สุดในสหรัฐ งานวิจัยอื่น ๆ ได้รายงานการกลายพันธุ์ที่โคดอนอื่น ๆ ซึ่งสัมพันธ์กับโรคอาร์พีรวมทั้ง Thr58Arg, Pro347Leu, Pro347Ser บวกกกับการหลุดหาย (deletion) ของยีน Ile-255
ในปี 2000 การกลายพันธุ์ที่มีน้อยของโคดอน 23 ได้ระบุว่าก่อโรคโดยสืบทอดทางพันธุกรรมแบบทายกรรมลักษณะเด่น และโปรลีนจะเปลี่ยนเป็นอะลานีน (alanine) แต่งานศึกษานี้ก็ได้แสดงว่า ความเสื่อมจอตาที่สัมพันธ์กับกลายพันธุ์นี้ค่อนข้างเบาทั้งโดยอาการและการลุกลาม อนึ่ง แอมพลิจูดที่วัดโดย electroretinography จะดีกว่าที่พบในการกลายพันธุ์ Pro23His ที่มีมากกว่า
การสืบทอดทางพันธุกรรมแบบทายกรรมลักษณะด้อยของโรคได้ระบุแล้วในยีนอย่างน้อย 45 ยีน การสืบทอดเช่นนี้หมายความว่า พ่อแม่ที่เป็นพาหะของยีนกลายพันธุ์ที่ก่อโรคโดยมีสองอัลลีล (diallelic) อาจมีลูกที่เกิดโรค
ส่วนการกลายพันธุ์ของยีนแบบ USH2A รู้ว่าเป็นเหตุของโรคแบบเกิดกับอาการอื่น (syndromic) ที่รู้จักว่า Usher's Syndrome โดยจะสืบทอดแบบทายกรรมลักษณะด้อย
การกลายพันธุ์ของ pre-mRNA splicing factors ถึง 4 แบบรู้แล้วว่าเป็นเหตุของโรคที่สืบทอดแบบทายกรรมลักษณะเด่น การกลายพันธุ์รวมทั้ง PRPF3 (PRPF3 ของมนุษย์ก็คือ HPRPF3 หรือ PRP3), PRPF8, PRPF31 และ PAP1 เพราะแฟกเตอร์มีการแสดงออกอย่างแพร่หลาย จึงเสนอว่าความบกพร่องของแฟกเตอร์ที่แพร่หลาย (คือโปรตีนที่แสดงออกทุกหนทุกแห่ง) เช่นนี้แต่เป็นเหตุของโรคภายในจอตาเท่านั้น เพราะโรด็อปซินในเซลล์รับแสงจำเป็นต้องอาศัยฤทธิ์ของแฟกเตอร์มากกว่าที่อื่น ๆ
การกลายพันธุ์ของยีน 6 ยีนก่อโรคที่สืบทอดแบบ X-linked โดยที่สามัญสุดจะเกิดที่ตำแหน่ง (loci) โดยเฉพาะ ๆ ของยีน RPGR และ RP2
รูปแบบของอาร์พีรวมทั้ง
| OMIM | ยีน | รูปแบบ |
|---|---|---|
| 180100 | RP1 | Retinitis pigmentosa-1 |
| 312600 | RP2 | Retinitis pigmentosa-2 |
| 300029 | RPGR | Retinitis pigmentosa-3 |
| 608133 | PRPH2 | Retinitis pigmentosa-7 |
| 180104 | RP9 | Retinitis pigmentosa-9 |
| 180105 | IMPDH1 | Retinitis pigmentosa-10 |
| 600138 | PRPF31 | Retinitis pigmentosa-11 |
| 600105 | CRB1 | Retinitis pigmentosa-12, ทายกรรมลักษณะด้อย |
| 600059 | PRPF8 | Retinitis pigmentosa-13 |
| 600132 | TULP1 | Retinitis pigmentosa-14 |
| 600852 | CA4 | Retinitis pigmentosa-17 |
| 601414 | HPRPF3 | Retinitis pigmentosa-18 |
| 601718 | ABCA4 | Retinitis pigmentosa-19 |
| 602772 | EYS | Retinitis pigmentosa-25 |
| 608380 | CERKL | Retinitis pigmentosa-26 |
| 607921 | FSCN2 | Retinitis pigmentosa-30 |
| 609923 | TOPORS | Retinitis pigmentosa-31 |
| 610359 | SNRNP200 | Retinitis pigmentosa 33 |
| 610282 | SEMA4A | Retinitis pigmentosa-35 |
| 610599 | PRCD | Retinitis pigmentosa-36 |
| 611131 | NR2E3 | Retinitis pigmentosa-37 |
| 268000 | MERTK | Retinitis pigmentosa-38 |
| 268000 | USH2A | Retinitis pigmentosa-39 |
| 612095 | PROM1 | Retinitis pigmentosa-41 |
| 612943 | KLHL7 | Retinitis pigmentosa-42 |
| 268000 | CNGB1 | Retinitis pigmentosa-45 |
| 613194 | BEST1 | Retinitis pigmentosa-50 |
| 613464 | TTC8 | Retinitis pigmentosa 51 |
| 613428 | C2orf71 | Retinitis pigmentosa 54 |
| 613575 | ARL6 | Retinitis pigmentosa 55 |
| 613617 | ZNF513 | Retinitis pigmentosa 58 |
| 613861 | DHDDS | Retinitis pigmentosa 59 |
| 613194 | BEST1 | Retinitis pigmentosa, concentric |
| 608133 | PRPH2 | Retinitis pigmentosa, digenic |
| 613341 | LRAT | Retinitis pigmentosa, juvenile |
| 268000 | SPATA7 | Retinitis pigmentosa, juvenile, ทายกรรมลักษณะด้อย |
| 268000 | CRX | Retinitis pigmentosa, late-onset dominant |
| 300455 | RPGR | Retinitis pigmentosa, X-linked, and sinorespiratory infections, อาจจหูหนวกหรือไม่ |
พยาธิสรีรภาพ

นักวิชาการได้จับคู่การทำงานระดับโมเลกุลที่บกพร่องหลายอย่างกับการกลายพันธุ์ของยีนเกี่ยวกับโรคแล้ว การกลายพันธุ์ของยีนโรด็อปซิน ซึ่งก่อโรคที่สืบทอดแบบทายกรรมลักษณะเด่นโดยมาก จะขัดการทำงานของโปรตีนอ็อปซินของเซลล์รูปแท่งซึ่งจำเป็นในการแปลข้อมูลแสงให้เป็นกระแสประสาท (phototransduction cascade) ของระบบประสาทกลาง ความผิดปกติในการทำงานของโรด็อปซิน ซึ่งเป็นหน่วยรับที่จับคู่กับจีโปรตีน (GPCR) จะจัดเป็นหมู่ ๆ (class) ขึ้นอยู่กับการพับตัวผิดปกติของโปรตีนและวิถีการทำงานที่ผิดปกติของโปรตีน
ฤทธิ์ของโปรตีนกลายพันธุ์หมู่หนึ่ง (Class I) จะเสียไปเพราะการกลายพันธุ์ของเบสนิวคลีโอไทด์ 1 เบส (point mutation) ภายในลำดับกรดอะมิโนที่เข้ารหัสโปรตีน และมีผลต่อการขนส่งโปรตีนเข้าไปที่ส่วนนอก (outer segment) ของเซลล์ ซึ่งเป็นส่วนที่ถ่ายโอนแสงเป็นกระแสประสาท อนึ่ง การพับตัวผิดปกติของโปรตีนกลายพันธุ์หมู่สอง (Class II) จะขัดการประกอบโปรตีนกับ 11-cis-retinal เพื่อสร้างส่วนกำเนิดสี (chromophore) ที่เหมาะสม การกลายพันธุ์อื่น ๆ ในยีนที่เข้ารหัสสารสีนี้จะมีผลต่อเสถียรภาพของโปรตีน, ทำลายบูรณภาพของ mRNA ในระยะ post-translational และมีผลต่ออัตราการก่อกัมมันต์ (activation rate) ของ transducin บวกกับอ็อปซินซึ่งเป็นโปรตีนนำแสง
การทดลองในสัตว์ตัวแบบแสดงว่า เซลล์ชั้น retinal pigment epithelium ของจอตาไม่ทำการฟาโกไซโทซิสต่อจานส่วนนอกของเซลล์รูปแท่ง (outer rod segment disc) ที่หลุดออกแล้ว เป็นการสะสมเศษส่วนนอกที่ละทิ้งแล้วอย่างผิดปกติ ในหนูที่มีการกลายพันธุ์ให้จอตาเสื่อมและสืบทอดแบบ homozygous recessive เซลล์รูปแท่งจะหยุดพัฒนาการแล้วเสื่อมก่อนที่เซลล์จะโตเต็มที่ อนึ่ง ได้พบความบกพร่องของเอนไซม์ cGMP-phosphodiesterase อีกด้วย ซึ่งทำให้ cyclic guanosine monophosphate (cGMP) สะสมในระดับที่เป็นพิษ
การวินิจฉัย
การวินิจฉัยโรคที่แม่นยำจะอาศัยประวัติความเสียหายที่แย่ลงอย่างต่อเนื่องของเซลล์รับแสง ซึ่งยืนยันด้วยการตรวจสอบต่าง ๆ รวมทั้งการตรวจลานสายตา (visual field test), การตรวจสายตา (visual acuity test), การถ่ายภาพจอตา, optical coherence imagery และ electroretinography (ERG)
การตรวจลานสายตาและการตรวจสายตาเทียบขนาดลานสายตาและความคมชัดของการเห็นกับค่าวัดของบุคคลที่มีสายตาสมบูรณ์ (20/20 vision) ลักษณะทางคลินิกที่บ่งโรครวมทั้งลานสายตาที่แคบลงและแย่ลง ๆ และสายตาที่มองชัดแย่ลง ๆ อนึ่ง การถ่ายภาพรังสีระนาบ (optical tomography) เช่นภาพ fundus optical coherence และ retinal optical coherence สามารถใช้ช่วยวินิจฉัยโรค การถ่ายภาพจอตาที่ขยายม่านตาแล้วช่วยยืนยันการเกิดรอยเปรอะดำคือ bony spicule ที่ก้นตา ซึ่งจะเกิดในระยะหลัง ๆ ของโรค เมื่อรวมกับภาพส่วนตัดด้วย optical coherence tomography ซึ่งให้ข้อมูลเกี่ยวกับความหนาของเซลล์รับแสง, สัณฐานของชั้นจอตา (retinal layer) และสรีรภาพของ retinal pigment epithelium ภาพถ่ายจอตาก็จะช่วยระบุความเคลื่อนไหวของโรคอาร์พี
แม้การตรวจลานสายตา การตรวจสายตา และภาพจอตาจะสนับสนุนการวินิจฉัยเป็นโรคอาร์พี แต่การทดสอบอื่น ๆ ก็จำเป็นเพื่อยืนยันลักษณะอื่น ๆ ของโรค Electroretinography (ERG) ยืนยันการวินิจฉัยเพราะตรวจสอบการทำงานของสายตาเนื่องกับการเสื่อมของเซลล์รับแสง และสามารถตรวจจับความผิดปกติทางสรีรภาพก่อนที่อาการจะปรากฏ เจ้าหน้าที่จะใช้เลนส์ที่เป็นอิเล็กโทรดเพื่อวัดการตอบสนองของเซลล์รับแสงต่อแสงกะพริบที่ฉายให้ดูในระดับความสว่างต่าง ๆ คนไข้ที่มีฟีโนไทป์ของโรคจะมีการตอบสนองทางไฟฟ้าที่ลดลงหรือช้าลงของเซลล์รูปแท่ง และอาจจะเซลล์รูปกรวยด้วย
เมื่อวินิจฉัย แพทย์อาจพิจารณาประวัติครอบครัวของคนไข้เพราะเป็นโรคที่อาจติดต่อทางกรรมพันธุ์ มียีนอย่างน้อย 35 ตำแหน่ง (locus) ที่เป็นเหตุของโรคแบบ "nonsyndromic" คืออาร์พีที่ไม่ใช่ผลของอีกโรคหนึ่ง หรือเป็นส่วนของกลุ่มอาการอื่น ๆ การกลายพันธุ์ของยีนที่ก่อโรคอาจระบุได้ด้วยการตรวจดีเอ็นเอ (DNA testing) ซึ่งมีใช้ทางคลินิกในบางประเทศ การกลายพันธุ์รวมทั้ง
สำหรับยีนอื่น ๆ (เช่น DHDDS) การตรวจดีเอ็นเอมีใช้ในการวิจัยเท่านั้น
โรคสามารถสืบทอดทางพันธุกรรมแบบทายกรรมลักษณะเด่น (autosomal dominant), ทายกรรมลักษณะด้อย (autosomal recessive) หรือ X-linked แบบ X-linked อาจเป็นแบบด้อย (recessive) ซึ่งโดยหลักมีผลต่อผู้ชายเท่านั้น หรือเป็นแบบเด่น (dominant) ซึ่งมีผลต่อทั้งชายหญิง แม้ชายจะมีอาการเบากว่า รูปแบบโรคที่เกิดจากยีนสองยีน (digenic) และจากไมโทคอนเดรียก็มีด้วย
ในบางประเทศ เจ้าหน้าที่อาจให้คำปรึกษาเกี่ยวกับปัญหาการสืบทอดทางพันธุกรรมในครอบครัว โดยขึ้นอยู่กับการวินิจฉัยที่ทำอย่างแม่นยำ การระบุวิธีสืบทอดทางพันธุกรรม และผลการตรวจดีเอ็นเอ
การรักษา
ปัจจุบันยังไม่มีวิธีรักษาโรคให้หาย แม้จะมีการตรวจสอบประสิทธิผลและความปลอดภัยของวิธีการรักษาต่าง ๆ อยู่ ประสิทธิภาพของอาหารเสริมต่าง ๆ เช่น วิตามินเอ, กรดไขมัน docosahexaenoic acid (DHA) และ Lutein เพื่อชะลอโรคยังเป็นเรื่องยังไม่ยุติ แต่ก็เป็นทางเลือกในการรักษา การทดลองทางคลินิกที่ตรวจสอบอุปกรณ์ฝังที่จอตา/เรตินาเทียม (optic prosthetic device), การบำบัดด้วยยีน (gene therapy) และการผ่าตัดเปลี่ยนจอตา (retinal sheet transplantation) เป็นเรื่องที่กำลังศึกษาเพื่อคืนการเห็นเป็นบางส่วนให้แก่คนไข้โรคอาร์พี
วิตามิน A ในรูปแบบ Palmitate
งานวิจัยปี 1993 ของศาสตราจารย์จักษุวิทยาผู้หนึ่งแห่งมหาวิทยาลัยฮาร์วาร์ดแสดงว่า คนไข้บางพวกสามารถชะลอความเสื่อมของเซลล์รูปแท่งโดยทานวิตามินเอในรูปแบบของ vitamin A palmitate ประมาณ 15,000 หน่วยสากล (เท่ากับ 4.5 มิลลิกรัม) ต่อวัน แม้จะไม่พบปัญหาความปลอดภัยเมื่อใช้วิตามินเอในระดับสูงในช่วงระยะงานวิจัย แต่ความปลอดภัยระยะยาวก็ยังไม่ชัดเจน เพราะการใช้วิตามินเอในระดับนี้อาจมีผลข้างเคียงหลายอย่างและจัดว่า สามารถก่อความผิดปกติทางพัฒนาการ (teratogenic) อนึ่ง บทความปริทัศน์ปี 2004 สรุปว่า
งานวิจัยและข้อแนะนำ (ของศาสตราจารย์ที่ว่า) นั้นยังมีข้อโต้แย้งอยู่ มีการเสนอว่า มีความจำเป็นต้องมีหลักฐานพิสูจน์ว่า มีประโยชน์ที่กว้างขวางยิ่งกว่านี้ ก่อนที่จะแนะนำการใช้วิตามินเอสำหรับอาร์พี
งานวิจัยของศาสตราจารย์คนเดียวกันปี 2007 แสดงว่าการใช้วิตามินเอที่เหมาะสมสำหรับคนไข้ในบางระยะของโรคบางรูปแบบ สามารถชะลอการตาบอดได้ถึง 10 ปี โดยลดความเสียหายที่เกิดขึ้นในอัตรา 10% ต่อปีให้เป็น 8.3% ต่อปี
เรตินาเทียม
อุปกรณ์ฝังในตา/เรตินาเทียม Argus II retinal prosthesis เป็นวิธีการรักษาโรคแรกที่ได้รับอนุมัติในเดือนกุมภาพันธ์ 2011 ปัจจุบันมีใช้รักษาในประเทศเยอรมนี ฝรั่งเศส อิตาลี และสหราชอาณาจักร ผลในระหว่างที่ได้จากคนไข้ 30 คนในการทดลองระยะยาวได้เผยแพร่ในปี 2012
ต่อมาปี 2013 องค์การอาหารและยาสหรัฐจึงได้อนุมัติให้ใช้การรักษานี้ในสหรัฐอเมริกา อุปกรณ์อาจช่วยคนไข้โรคอาร์พีผู้ใหญ่ที่เสียการเห็นรูปร่างและการเคลื่อนไหวเพื่อให้สามารถใช้ชีวิตประจำวันและช่วยเหลือตัวเองได้ดีขึ้น แม้คนไข้จะเห็นได้ชัดเจนที่สุดในระดับ 20/1260 เท่านั้น ซึ่งยังจัดว่าบอดอยู่ทั้งโดยองค์การอนามัยโลกและประเทศสหรัฐอเมริกา ในเดือนมิถุนายน 2013 รพ. 12 แห่งในสหรัฐประกาศว่าจะเริ่มรับคนไข้โรคอาร์พีที่สนใจจะใช้อุปกรณ์ที่จะเริ่มวางตลาดในปลายปีนั้น ค่าใช้จ่ายสำหรับอุปกรณ์อยู่ที่ 150,000 ดอลลาร์สหรัฐ (ประมาณ 4.7 ล้านบาท) โดยไม่รวมค่าผ่าตัดหรือค่าฝึกใช้อุปกรณ์
ส่วนอุปกรณ์ Alpha-IMS เป็นไมโครชิปบันทึกภาพที่สามารถผ่าตัดฝังใต้รอยบุ๋มจอตา (fovea) แต่เพื่อศึกษาว่าอุปกรณ์สามารถทำให้เห็นดีขึ้นหรือไม่ ผู้ผลิตจำต้องแสดงความปลอดภัยของอุปกรณ์ก่อนทำการทดลองทางคลินิกและอนุมัติให้วางตลาด โดยค่าอุปกรณ์และการผ่าตัดจะอยู่ที่ประมาณ 100,000 ยูโร (ประมาณ 4.1 ล้านบาท)
การบำบัดด้วยยีน (gene therapy)
ส่วนเป้าหมายของการศึกษาเรื่องการบำบัดด้วยยีน (gene therapy) ก็คือเพื่อเสริมเซลล์จอตาอันแสดงออกยีนที่กลายพันธุ์ซึ่งสัมพันธ์กับฟีโนไทป์ของโรคด้วยยีนที่ดี เพื่อให้เซลล์รับแสงสามารถซ่อมแซมตนเองและทำงานได้อย่างถูกต้องด้วยข้อมูลที่ได้จากยีนที่ดี การทดลองทางคลินิกที่ใส่ยีน RPE65 ปกติในจอตาที่แสดงออกฟีโนไทป์โรคอาร์พีแบบ LCA2 (Leber's congenital amaurosis) พบว่าทำให้เห็นดีขึ้นได้เล็กน้อย แต่เซลล์รับแสงก็ยังเสื่อมในอัตราปกติของโรค การบำบัดด้วยยีนน่าจะช่วยรักษาเซลล์จอตาที่ยังดีอยู่ แต่ไม่สามารถซ่อมความเสียหายต่อเซลล์รับแสงที่สั่งสมมาก่อน ๆ ได้ โดยทฤษฎีแล้ว การตอบสนองรับการบำบัดด้วยยีนน่าจะดีต่อคนไข้วัยเยาว์ที่โรคมีระยะการดำเนินสั้นที่สุด เพราะมีโอกาสช่วยเซลล์ให้พ้นโรคด้วยยีนที่ใส่เข้าไปมากที่สุด
ในปี 2017 องค์การอาหารและยาสหรัฐอนุมัติการบำบัดด้วยีนที่เรียกว่า Luxturna เพื่อรักษาคนไข้ที่จอตาฝ่อ (retinal dystrophy) เนื่องกับการกลายพันธุ์ของยีน RPE65 ค่ารักษาอยู่ที่ 850,000 ดอลลาร์สหรัฐ (ประมาณ 27.5 ล้านบาท) สำหรับตาทั้งสองข้างเมื่อต้นปี 2018
พยากรณ์โรค
เพราะโรคแย่ลงเรื่อย ๆ และไม่มีวิธีการรักษา คนไข้จึงมีพยากรณ์โรคที่ไม่ค่อยดี แม้ปกติตาจะไม่บอดโดยสิ้นเชิง สายตาและลานสายตาของคนไข้จะแย่ลงเรื่อย ๆ เริ่มจากการเสื่อมของเซลล์รูปแท่งและต่อมาเซลล์รูปกรวย การรักษาที่เป็นไปได้ก็ยังอยู่ในระยะวิจัยหรือการทดลองทางคลินิก อย่างไรก็ดี การรักษาด้วยการคืนสภาพสายตาดูเหมือนจะมีอนาคตที่แจ่มใส
งานศึกษาได้แสดงว่า เด็กที่มีจีโนไทป์ของโรคจะได้ประโยชน์จากการปรึกษากับแพทย์ก่อนจะมีโรค เพื่อเตรียมตัวรับมือปัญหาทางตาและทางสังคมเนื่องกับสายตาที่แย่ลงเรื่อย ๆ แม้ปัญหาทางจิตใจจะดีขึ้นเมื่อทำเช่นนี้ แต่ผลทางตาและการแย่ลงของโรคโดยมากจะขึ้นอยู่กับอายุที่อาการเริ่มปรากฏและอัตราการเสื่อมของเซลล์รับแสง ไม่ใช่การมีโอกาสไปหาแพทย์ อุปกรณ์ปรับสายตาและการบำบัดตาที่ปรับให้เหมาะกับตนซึ่งทำกับผู้เชี่ยวชาญในเรื่องการมองเห็นได้น้อย อาจช่วยคนไข้แก้ปัญหาการมองเห็น คือช่วยให้มองด้วยลานสายตาที่เหลืออยู่ได้ดีที่สุด กลุ่มสนับสนุน, ประกันเกี่ยวกับสายตา และการปรับวิถีชีวิต อาจเป็นประโยชน์ในการรับมือกับการเห็นที่ลดลง
วิทยาการระบาด
โรคอาร์พีเป็นเหตุชั้นนำของความตาบอดที่สืบทอดทางพันธุกรรม คนประมาณ 1 ใน 4,000 คนจะมีโรคแบบไม่เกิดร่วมกับความผิดปกติอื่น ๆ (non-syndromic) ในช่วงชีวิต ประเมินว่ามีคน 1.5 ล้านคนทั่วโลกที่มีโรค โรคที่เริ่มเกิดเร็วจะเกิดภายในไม่กี่ปีแรกของชีวิตและปกติจะสัมพันธ์กับความผิดปกติอื่น ๆ ในขณะที่โรคที่เริ่มเกิดช้าจะปรากฏในวัยผู้ใหญ่ต้น ๆ จนถึงวัยกลางคน
โรคที่สืบทอดแบบทายกรรมลักษณะเด่นและทายกรรมลักษณะด้อยจะมีทั้งในชายหญิงเท่า ๆ กัน แต่โรคที่สืบทอดแบบ X-linked ซึ่งมีน้อยกว่าจะมีผลต่อชายที่ได้รับยีนกลายพันธุ์ แต่หญิงจะเป็นพาหะของโรคโดยไม่มีอาการ รูปแบบนี้จัดว่ารุนแรง และปกติจะทำให้ตาบอดสนิทในช่วงท้าย ๆ ส่วนในกรณีที่มีน้อย โรคที่สืบทอดแบบ X-linked เป็นลักษณะเด่นจะมีผลต่อทั้งชายหญิงเท่า ๆ กัน
เนื่องจากการสืบทอดทางพันธุกรรมของโรค กลุ่มประชากรที่แยกอยู่ต่างหากจะเกิดโรคบ่อยกว่าหรือมีความชุกโรคที่สูงขึ้นโดยมีการกลายพันธุ์ของยีนโดยเฉพาะ ๆ ที่ก่อโรค การกลายพันธุ์ไม่ว่าจะเป็นแบบที่มีอยู่แล้วหรือเกิดขึ้นใหม่ซึ่งทำให้เซลล์รับแสงรูปแท่งเสื่อมในโรคอาร์พีจะสืบทอดมาตามครอบครัว ดังนั้น จึงทำให้โรคเกิดขึ้นมากในภูมิภาคบางส่วนโดยปรากฏประวัติในครอบครัว มีงานศึกษาหลายงานที่ได้ทำเพื่อระบุความชุกโรคต่าง ๆ ในรัฐเมน (สหรัฐ) เมืองเบอร์มิงแฮม (สหราชอาณาจักร) ประเทศสวิตเซอร์แลนด์ (1 คนใน 7,000 คน) ประเทศเดนมาร์ก (1 คนใน 2,500 คน) และประเทศนอร์เวย์ คนพื้นเมืองอเมริกันชาวนาวาโจ (Navajo) มีระดับการสืบทอดโรคที่สูงขึ้นเช่นกัน โดยประเมินว่ามีผลต่อคน 1 คนทุก ๆ 1,878 คน โรคอาร์พีแม้จะสืบทอดตามกรรมพันธุ์ในครอบครัว แต่ก็จัดว่าไม่จำกัดเฉพาะกลุ่มบุคคล ๆ เพราะมีผลต่อประชากรทั่วโลก
งานวิจัย
การรักษาในอนาคตอาจรวมการผ่าตัดเปลี่ยนจอตา การใช้จอตาเทียม การบำบัดด้วยยีน (gene therapy) การบำบัดด้วยเซลล์ต้นกำเนิด อาหารเสริม และ/หรือยารักษา
ในปี 2006 นักวิจัยชาวอังกฤษได้ปลูกเซลล์ต้นกำเนิดที่ได้พัฒนาการมาจนถึงระยะสุดท้ายพร้อมที่จะกลายเป็นเซลล์รับแสง เข้าในตาของหนูที่ได้เปลี่ยนพันธุกรรมให้มีอาการเลียนโรคอาร์พีและโรคจุดภาพชัดของจอตาเสื่อมของมนุษย์ แล้วพบว่า เซลล์รับแสงเช่นนี้ได้พัฒนาการแล้วเชื่อมต่อกับเซลล์ประสาทในจอตาของหนู ซึ่งเป็นขั้นตอนสำคัญในการคืนสภาพสายตา เพราะก่อนหน้านี้ได้เชื่อว่า จอตาที่พัฒนาสมบูรณ์แล้วไม่สามารถฟื้นฟูซ่อมแซมได้ งานวิจัยนี้อาจทำให้สามารถปลูกฝังเซลล์เช่นนี้ในมนุษย์ในอนาคตได้
ในปี 2008 นักวิทยาศาสตร์ชาวญี่ปุ่นที่สถาบันชีววิทยาศาสตร์โอซากะ (Osaka Bioscience Institute) ได้ค้นพบโปรตีนที่ตั้งชื่อว่า Pikachurin ซึ่งพวกเขาเชื่อว่าอาจนำไปสู่วิธีการรักษาโรคอาร์พี
ในปี 2008 มีความพยายามที่จะเชื่อมการแสดงออกของยีน FAM46A กับโรค
ในปี 2010 การรักษาด้วยยีนวิธีหนึ่งดูเหมือนจะได้ผลในหนู
ในปี 2012 นักวิทยาศาสตร์ที่ศูนย์การแพทย์มหาวิทยาลัยโคลัมเบียได้แสดงกับสัตว์ตัวแบบว่า การรักษาด้วยยีน (gene therapy) และการรักษาด้วยเซลล์ต้นกำเนิดแบบ induced pluripotent stem cell อาจเป็นทางเลือกที่เป็นไปได้เพื่อรักษาโรคในอนาคต
ในปี 2012 นักวิทยาศาสตร์ที่สถาบันจักษุแบสคอมปาลเมอร์ (Bascom Palmer Eye Institute) แห่งมหาวิทยาลัยไมแอมี ได้แสดงการป้องกันรักษาเซลล์รับแสงในสัตว์ตัวแบบ เมื่อฉีด mesencephalic astrocyte-derived neurotrophic factor (MANF) เข้าไปในตา
ในปี 2014 นักวิจัยที่มหาวิทยาลัยแคลิฟอร์เนีย เบิร์กลีย์ได้คืนสภาพการเห็นของหนูตาบอดโดยใช้ photoswitch ที่เริ่มการทำงานของ retinal ganglion cell ในสัตว์ที่มีเซลล์รับแสง (ทั้งรูปแท่งและรูปกรวย) เสียหาย
งานศึกษาปี 2015 ที่ศูนย์การแพทย์ซีดาร์ส-ซีนาย (Cedars-Sinai Medical Center) ในนครลอสแอนเจลิสแสดงว่า ยีน CRISPR/Cas9 สามารถใช้รักษาหนูที่มีโรคซึ่งสืบทอดแบบทายกรรมลักษณะเด่น
ในปี 2016 บริษัท RetroSense Therapeutics มีแผนจะฉีดไวรัสที่มีดีเอ็นเอของสาหร่ายไวแสงเข้าไปในตาของคนตาบอดเนื่องกับโรคอาร์พีหลายคน โดยเป็นโครงการที่ได้รับอนุมัติจากองค์การอาหารและยาสหรัฐ ถ้าประสบความสำเร็จ ก็จะเห็นเป็นขาวดำ
ดูเพิ่ม
เชิงอรรถ
แหล่งข้อมูลอื่น
| การจำแนกโรค | |
|---|---|
| ทรัพยากรภายนอก |