
| โรคฮันติงตัน (Huntington's disease) | |
|---|---|
| ชื่ออื่น | Huntington's chorea, Saint Vitus' dance |
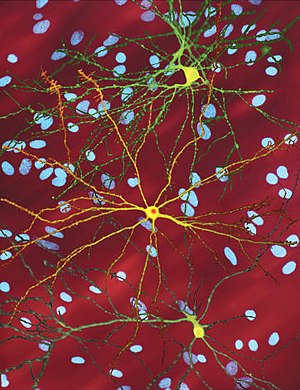 | |
| ภาพจากกล้องจุลทรรศน์แสดงให้เห็น medium spiny neuron (สีเหลือง) ซึ่งมีอินคลูชันบอดี้ในนิวเคลียส เป็นลักษณะบ่งบอกว่ากำลังมีการดำเนินของโรคอยู่ ภาพนี้มีความกว้าง 360 ไมโครเมตร | |
| สาขาวิชา | Neurology |
| อาการ | Problems with mood, mental abilities, coordination, jerky body movements |
| ภาวะแทรกซ้อน | Pneumonia, heart disease, physical injury from falls, suicide |
| การตั้งต้น | 30–50 ปี |
| ระยะดำเนินโรค | ระยะยาว |
| สาเหตุ | Genetic (inherited or new mutation) |
| วิธีวินิจฉัย | Genetic testing |
| โรคอื่นที่คล้ายกัน | Sydenham's chorea, benign hereditary chorea, lupus, paraneoplastic syndrome, Wilson's disease |
| การรักษา | Supportive care |
| ยา | Tetrabenazine |
| พยากรณ์โรค | 15–20 ปีจากการวินิจฉัย |
| ความชุก | 4–15 in 100,000 (European descent) |
| การเสียชีวิต | 40 (2019) |
โรคฮันติงตัน (อังกฤษ: Huntington's disease / chorea / disorder) เป็นโรคทางพันธุกรรมโรคหนึ่งที่ทำให้เกิดการเสื่อมของระบบประสาท ส่งผลต่อการควบคุมการประสานงานของกล้ามเนื้อ ทำให้สติปัญญาเสื่อมถอย และนำไปสู่ภาวะสมองเสื่อมได้ ส่วนใหญ่ปรากฏอาการในช่วงวัยกลางคน เป็นโรคที่เป็นสาเหตุทางพันธุกรรมที่พบบ่อยที่สุดของภาวะการเคลื่อนไหวผิดปกติที่เรียกว่าโคเรีย และพบในคนเชื้อชาติยุโรปตะวันตกมากกว่าเอเชียหรือแอฟริกา เกิดจากการกลายพันธุ์ในยีนฮันติงตินยีนหนึ่งในสองยีนในร่างกาย ซึ่งมีการถ่ายทอดลักษณะแบบลักษณะเด่น ดังนั้นทายาทของผู้ป่วยโรคนี้มีโอกาสได้รับถ่ายทอดโรคนี้มา 50% ในบางกรณีที่ทั้งบิดาและมารดามียีนที่เป็นโรคคนละหนึ่งในสองยีน ทายาทจะมีโอกาสเป็นโรค 75% และหากมีบิดาหรือมารดามียีนที่เป็นโรคสองยีน ทายาทก็จะมีโอกาสติดโรค 100% อาการทางกายของโรคฮันติงตันอาจเริ่มปรากฏได้ตั้งแต่วัยทารกไปจนถึงวัยชรา แต่ส่วนใหญ่จะปรากฏในช่วงอายุ 35-44 ปี ผู้ป่วยประมาณ 6% เริ่มมีอาการตั้งแต่ก่อนอายุ 21 ปี โดยมีกลุ่มอาการกล้ามเนื้อเกร็งและเริ่มต้นเคลื่อนไหวลำบาก ซึ่งผู้ป่วยกลุ่มนี้อาการจะทรุดลงรวดเร็วเกือบทุกคน โรคฮันติงตันที่แสดงอาการเช่นนี้เรียกว่าโรคฮันติงตันวัยเด็ก (อังกฤษ: juvenile) หรือกล้ามเนื้อเกร็งเคลื่อนไหวลำบาก (อังกฤษ: akinetic-rigid) หรือชนิดเวสท์ฟาล (อังกฤษ: Westphal varient)
ยีนฮันติงตินเก็บข้อมูลทางพันธุกรรมสำหรับสร้างโปรตีนชื่อว่าฮันติงติน การกลายพันธุ์ของยีนฮันฮิงตินนี้ทำให้มีการสร้างโปรตีนซึ่งผิดปกติออกมา ซึ่งทำให้เกิดการเสื่อมลงอย่างช้าๆ ในบางบริเวณของสมอง กลไกของการเสื่อมที่เกิดจากการสร้างโปรตีนนี้ยังไม่เป็นที่เข้าใจอย่างสมบูรณ์ ปัจจุบันมีการตรวจทางพันธุกรรมที่สามารถพบการกลายพันธุ์ของยีนนี้ได้ในทุกระยะของการเจริญ รวมถึงก่อนเริ่มมีอาการด้วย ทำให้เป็นประเด็นถกเถียงทางจริยธรรมว่าผู้สงสัยเป็นโรคนั้นควรได้รับการตรวจหายีนก่อโรคเมื่ออายุเท่าไรจึงจะเหมาะสม สิทธิของบิดามารดาในการตรวจหาโรคในบุตร และการรักษาความลับของผลตรวจนั้นๆ มีการพัฒนาการให้คำปรึกษาทางพันธุกรรมเพื่อที่จะให้ข้อมูลและช่วยเหลือผู้ที่ต้องการตรวจหาโรคนี้ และกลายเป็นต้นแบบในการให้คำปรึกษาทางพันธุกรรมสำหรับโรคทางพันธุกรรมอื่นๆ ที่มีลักษณะการถ่ายทอดแบบลักษณะเด่นเช่นเดียวกัน
อาการของโรคในผู้ป่วยแต่ละคนอาจแตกต่างกันได้มาก บางครั้งผู้ป่วยในครอบครัวเดียวกันก็อาจมีอาการแตกต่างกันอย่างมากได้ แต่ผู้ป่วยส่วนใหญ่จะมีอาการแย่ลงเรื่อยๆ อาการแรกเริ่มโดยทั่วไปจะเป็นอาการของกล้ามเนื้อเสียการประสานงานและเดินไม่มั่นคง เมื่อโรคดำเนินไปอาการกล้ามเนื้อเสียการประสานงานและการเคลื่อนไหวผิดปกติจะเด่นชัดขึ้น พร้อมๆ กับที่มีกรสูญเสียความสามารถทางจิตใจ สติปัญญา และพฤติกรรม รวมทั้งอาจมีอาการทางจิตเวชได้ด้วย ความสามารถทางกายจะค่อยๆ เสื่อมลงจนในที่สุดการเคลื่อนไหวง่ายๆ ก็อาจกลายเป็นเรื่องยาก ความสามารถทางจิตใจอาจเสื่อมลงจนเข้าสู่ภาวะสมองเสื่อม อาจเกิดภาวะแทรกซ้อนต่างๆ เช่น ปอดบวม โรคหัวใจ อุบัติเหตุจากการพลัดตกหกล้ม จนทำให้อายุขัยสั้นลงอยู่ที่ประมาณ 20 ปีหลังเริ่มมีอาการ ปัจจุบันโรคฮันติงตันยังไม่มีวิธีรักษา ผู้ป่วยระยะท้ายของโรคจำเป็นต้องได้รับการดูแลช่วยเหลือตลอดเวลา แต่ระยะหลังเริ่มมีการรักษาใหม่ๆ ที่บรรเทาอาการบางอย่างของโรคได้
มีการก่อตั้งองค์กรช่วยเหลือตนเองสำหรับผู้ป่วยโรคฮันติงตันเป็นครั้งแรกในช่วงคริสต์ทศวรรษ 1960 และเพิ่มจำนวนมากขึ้นเรื่อยๆ ซึ่งองค์กรเหล่านี้จำนวนมากดำเนินการในด้านการให้ข้อมูลและสร้างความตื่นตัวให้กับสังคม ให้ความช่วยเหลือกับผู้ป่วยและครอบครัว และสนับสนุนการศึกษาวิจัยที่เกี่ยวข้อง มูลนิธิโรคที่สืบทอดทางพันธุกรรม (The Hereditary Disease Foundation) เป็นกลุ่มศึกษาวิจัยกลุ่มหนึ่งที่แตกยอดออกมาจากองค์กรให้ความช่วยเหลือที่ก่อตั้งขึ้นเป็นองค์กรแรกๆ ได้มีบทบาทในการค้นหายีนก่อโรคใน ค.ศ. 1993 ซึ่งหลังจากการค้นพบนี้แล้วก็มีการค้นพบสำคัญๆ ออกมาอย่างต่อเนื่องทุกๆ 2-3 ปี ทำให้องค์ความรู้เกี่ยวกับโรคฮันติงตันมีการพัฒนาอย่างต่อเนื่อง แนวทางการศึกษาวิจัยในปัจจุบันส่วนหนึ่งเน้นศึกษาหากลไกที่แท้จริงของการเกิดโรค พัฒนาสัตว์ทดลองที่เหมาะสมกับการศึกษาวิจัย ดำเนินการวิจัยเชิงทดลองทางคลินิกของยาที่รักษาอาการหรือชะลอการดำเนินโรค และศึกษาวิธีการรักษาใหม่ๆ อย่างเช่นการนำเซลล์ต้นกำเนิดมาใช้ในการรักษา โดยมีเป้าหมายเพื่อซ่อมแซมความเสียหายที่เกิดจากโรคนี้
อาการและอาการแสดง
ส่วนใหญ่ผู้ป่วยโรคฮันติงตันมักเริ่มมีอาการในช่วงอายุ 35-44 ปี แต่ก็สามารถเริ่มมีอาการได้ในทุกช่วงอายุตั้งแต่วัยทารกไปจนถึงวัยชรา ในระยะแรกๆ ของโรคผู้ป่วยอาจมีการเปลี่ยนแปลงเล็กน้อยทางด้านบุคลิกภาพ สติปัญญา และทักษะทางกาย ส่วนใหญ่อาการที่มักได้รับการสังเกตเป็นอันดับแรกๆ คืออาการทางกาย เนื่องจากอาการทางสติปัญญาและทางจิตใจมักไม่เป็นรุนแรงมากจนเป็นที่สังเกตได้ในระยะแรกๆ ของโรค เมื่อโรคดำเนินไปมากขึ้นผู้ป่วยโรคฮันติงตันเกือบทุกคนจะมีอาการทางกายที่คล้ายคลึงกัน แต่อาการแรกเริ่ม ช่วงเวลาที่เริ่มมีอาการ ความเร็วในการดำเนินโรค และระดับของความบกพร่องในด้านสติปัญญาและจิตใจนั้นแตกต่างกันมากในผู้ป่วยแต่ละคน
อาการแรกเริ่มที่มีลักษณะเฉพาะตัวคือ การมีการเคลื่อนไหวผิดปกติแบบกระตุก เป็นขึ้นโดยฉับพลันไม่มีสัญญาณเตือนล่วงหน้าและควบคุมไม่ได้ เรียกว่าโคเรีย อาการแรกเริ่มของโคเรียนี้อาจเป็นเพียงความรู้สึกอยู่ไม่สุขที่ไม่มีลักกษณะเฉพาะใดๆ อาจเป็นการเคลื่อนไหวที่ไม่มีความหมายพิเศษเกิดขึ้นเป็นช่วงสั้นๆ โดยไม่ได้ตั้งใจ อาจเป็นการที่ไม่สามารถควบคุมการเคลื่อนไหวให้แม่นยำได้ หรืออาจมี saccadic eye movement ที่ช้าลง อาการผิดปกติของระบบกล้ามเนื้อนี้ส่วนใหญ่แล้วจะพบเกิดขึ้นก่อนอาการของความผิดปกติของการเคลื่อนไหวที่เห็นได้ชัดเจนอื่นๆ เป็นเวลาอย่างน้อย 3 ปี โดยอาการที่ชัดเจนเช่นอาการกล้ามเนื้อแข็งเกร็ง การเคลื่อนไหวบิดงอ หรือมีกล้ามเนื้อแข็งเกร็งในท่าผิดปกติ จะพบได้เมื่อโรคดำเนินไปมากขึ้น อาการแสดงเหล่านี้เกิดจากการที่สมองส่วนที่ควบคุมการเคลื่อนไหวได้รับความเสียหาย การทำงานของระบบควบคุมการเคลื่อนไหวจะค่อยๆ เสื่อมลงเรื่อยๆ จนถึงที่สุดแล้วการเคลื่อนไหวใดๆ ที่ปกติจะต้องอยู่ภายใต้การควบคุมของจิตใจ จะได้รับผลกระทบไปทั้งหมด ผลที่มักพบตามมาคืออาการทรงตัวลำบาก แสดงสีหน้าผิดปกติ เคี้ยวลำบาก กลืนลำบาก และพูดลำบาก อาการกินลำบากเหล่านี้มักทำให้ผู้ป่วยมีน้ำหนักลดและมีภาวะขาดสารอาหารตามมา อาการอื่นที่พบได้เช่นความผิดปกติของการนอนหลับ โรคฮันติงตันชนิดวัยเด็กจะมีอาการแตกต่างออกไปโดยจะมีการดำเนินโรคที่เร็วกว่ามาก อาจมีอาการโคเรียเพียงแค่ช่วงสั้นๆ หรือไม่มีเลย และเริ่มปรากฏอาการกล้ามเนื้อแข็งเกร็งอย่างรวดเร็ว โรคฮันติงตันชนิดนี้ยังอาจพบอาการชักได้บ่อยอีกด้วย
| กระสับกระส่าย | 38–73% |
| ไร้อารมณ์ | 34–76% |
| วิตกกังวล | 34–61% |
| ซึมเศร้า | 33–69% |
| ย้ำคิดย้ำทำ | 10–52% |
| จิตเภท | 3–11% |
ระดับสติปัญญาจะค่อยๆ เสื่อมถอยลง โดยเฉพาะความสามารถด้านการควบคุมที่ใช้สติปัญญา เช่น การวางแผน ความยืดหยุ่นทางสติปัญญา ความคิดเชิงนามธรรม การทำความเข้าใจและปฏิบัติตามกฎระเบียบ การแสดงพฤติกรรมที่เหมาะสม และการยับยั้งพฤติกรรมที่ไม่เหมาะสม เมื่อโรคดำเนินไปมากขึ้นจะเริ่มมีความเสียหายของความจำ มีรายงานของระดับความเสียหายของความทรงจำตั้งแต่การสูญเสียความจำระยะสั้นไปจนถึงการสูญเสียความทรงจำระยะยาว รวมไปถึงสูญเสีย episodic memory, procedural memory (ความสามารถในการทำกิจกรรมต่างๆ) และความทรงจำที่ใช้ในการทำงาน ปัญหาทางสติปัญญาจะค่อยๆ เป็นมากขึ้นๆ สุดท้ายแล้วอาจเป็นถึงภาวะสมองเสื่อม แบบแผนของการเสื่อมเช่นนี้ถูกเรียกว่า subcortical dementia syndrome (กลุ่มอาการสมองเสื่อมจากใต้เปลือกสมอง) เพื่อแยกจากอาการที่เกิดจากสมองเสื่อมจากเปลือกสมองของโรคอื่นๆ เช่น โรคอัลไซเมอร์
อาการทางจิตที่มีการรายงานได้แก่ วิตกกังวล ซึมเศร้า ไม่แสดงอารมณ์ เห็นแก่ตัว (egocentric) ก้าวร้าว และย้ำคิดย้ำทำ ซึ่งอาการย้ำคิดย้ำทำนี้อาจนำไปสู่อาการเสพติดอื่นๆ เช่น ติดเหล้า ติดการพนัน และติดเพศสัมพันธ์ นอกจากนี้ยังมีรายงานถึงการไม่สามารถสังเกตว่าอีกฝ่ายกำลังมีความรู้สึกทางลบอยู่อีกด้วยความชุกของอาการเหล่านี้มีความแตกต่างกันมากระหว่างการศึกษาวิจัยแต่ละครั้ง โดยรวมแล้วประมาณว่ามีความชุกตลอดชีวิตของความผิดปกติทางจิตอยู่ที่ 33-76% สำหรับผู้ป่วยหลายคนและบางครอบครัวอาการทางจิตเหล่านี้เป็นหนึ่งในอาการที่ส่งผลมากที่สุด มักมีผลต่อการดำเนินชีวิตประจำวัน และบ่อยครั้งเป็นสาเหตุของการที่ทำให้ไม่สามารถออกจากบ้านหรือสถานพยาบาลได้ นอกจากนี้ผู้ป่วยยังมีความคิดฆ่าตัวตายและการลงมือกระทำการฆ่าตัวตายมากกว่าคนทั่วไปอีกด้วย
ฮันติงตินที่ผิดปกติไม่ได้ส่งผลต่อสมองอย่างเดียวแต่ส่งผลต่อร่างกายส่วนอื่นซึ่งอาศัยการทำงานของฮันติงตินด้วย ความผิดปกติที่พบ เช่น กล้ามเนื้อฝ่อลีบ หัวใจวาย ความทนกลูโคสบกพร่อง (impaired glucose tolerance) น้ำหนักลด กระดูกพรุน และอัณฑะฝ่อ
พันธุศาสตร์
มนุษย์ทุกคนมียีนฮันติงติน (HTT) อยู่เป็นปกติ ยีนนี้ทำหน้าที่สร้างโปรตีนฮันติงติน (Htt) ส่วนหนึ่งของยีนฮันฮิงตินนี้เป็นบริเวณที่มีคู่เบสลำดับซ้ำกันเป็นชุดสามเรียกว่า trinucleotide repeat ซึ่งมีความยาวแตกต่างกันไปในแต่ละคนและอาจมีการเปลี่ยนแปลงของความยาวได้เมื่อมีการถ่ายทอดจากรุ่นสู่รุ่น เมื่อบริเวณนี้ของยีนมีความยาวมากถึงค่าหนึ่งจะทำให้การสร้างโปรตีนผิดปกติไป โดยสร้างเป็นโปรตีนฮันติงตินที่มีการกลายพันธุ์ (mutant Huntingtin protein) หรือ mHtt ซึ่งเป็นโปรตีนที่มีคุณสมบัติแตกต่างกับ Htt ปกติ ทำให้เกิดการเปลี่ยนแปลงที่นำไปสู่การเป็นโรคฮันติงตัน การกลายพันธุ์ที่เกิดขึ้นในโรคฮันติงตันนั้นมีการถ่ายทอดแบบลักษณะเด่นและมีเพเนแทรนซ์เกือบ 100% (หมายความว่าเมื่อได้รับยีนก่อโรคมาแล้ว จะมีโอกาสที่โรคจะแสดงอาการเกือบ 100%) โดยการกลายพันธุ์ของยีน HTT ยีนใดยีนหนึ่งในสองยีน ก็ทำให้เป็นโรคได้ทั้งสิ้น การถ่ายทอดของโรคฮันติงตันไม่เกี่ยวข้องกับเพศ แต่ความยาวของบริเวณลำดับซ้ำในยีนซึ่งขึ้นตรงกับความรุนแรงของโรคด้วยนั้นมีความสัมพันธ์กับการได้รับยีนที่เป็นโรคมาจากฝ่ายบิดาหรือมารดา
ลักษณะการกลายพันธุ์
โรคฮันติงตันเป็นโรคในกลุ่มที่มีความผิดปกติของจำนวนลำดับซ้ำชุดสามของคู่เบสในดีเอ็นเอโรคหนึ่งจากจำนวนหลายๆ โรค ซึ่งโรคในกลุ่มนี้เกิดจากการที่จำนวนลำดับซ้ำชุดสามของคู่เบสในดีเอ็นเอมีจำนวนมากกว่าค่าปกติ ยีน HTT อยู่ในแขนสั้นของโครโมโซมคู่ที่ 4 ที่ตำแหน่ง 4p16.3 ซึ่งมีลำดับของเบสสามตัวซ้ำกัน ได้แก่ ไซโตซีน-อะดีนีน-กวานีน (CAG) ซึ่งเรียงต่อกันเรื่อยๆ เป็น CAGCAGCAG... ลักษณะนี้เรียกว่า trinucleotide repeat ซึ่ง CAG เป็นรหัสที่จะถูกถอดรหัสออกมาเป็นกรดอะมิโนกลูตามีน ดังนั้นลำดับซ้ำชุดสามของ CAG จะถูกถอดรหัสออกมาเป็นสายโซ่โปรตีนกลูตามีนต่อกัน เรียกว่าสายโพลีกลูตามีน หรือสาย polyQ ดังนั้นบริเวณที่มีการซ้ำกันของลำดับดีเอ็นเอนี้จึงถูกเรียกว่า PolyQ region (บริเวณ PolyQ)
| จำนวนชุดซ้ำ | ประเภท | ลักษณะของโรค |
|---|---|---|
| <28 | ปกติ | ไม่เป็นโรค |
| 28–35 | ปานกลาง | ไม่เป็นโรค |
| 36–40 | มีเพเนแทรนซ์ไม่ถึง 100% | อาจเป็นโรค |
| >40 | มีเพเนแทรนซ์ 100% | เป็นโรค |
คนปกติจะมีกลูตามีนซ้ำกันใน polyQ region ไม่เกิน 36 ชุด ซึ่งจะทำให้มีการถอดรหัสออกมาเป็นโปรตีนฮันติงตินชนิดปกติ คือ Htt ออกมาอยู่ในซัยโตพลาสซึมเพื่อทำหน้าที่ต่อไป แต่ถ้ามีซ้ำกันตั้งแต่ 36 ชุดขึ้นไปจะทำให้มีการสร้างโปรตีนผิดปกติคือ mHtt ซึ่งทำให้เซลล์ประสาทหรือนิวรอนบางชนิดเสื่อมเร็วผิดปกติ สมองแต่ละบริเวณมีเซลล์ประสาทที่ต้องใช้โปรตีนฮันติงตินเช่นนี้อยู่มากน้อยไม่เท่ากัน ทำให้ได้รับผลกระทบจากความผิดปกติของการสร้างโปรตีนฮันติงตินไม่เท่ากัน โดยทั่วไปแล้วจำนวนชุดซ้ำของ CAG เป็นสิ่งที่ส่งผลต่อการเกิดโรคฮันติงตันในแง่ของอายุที่จะเริ่มมีอาการ ซึ่งถือว่ามีผล 60% ความแปรปรวนที่นอกเหนือจากนี้ขึ้นกับปัจจัยสิ่งแวดล้อมและยีนอื่นๆ ที่ส่งผลต่อการดำเนินโรคของโรคฮันติงตัน การมีจำนวนชุด CAG ซ้ำ 36-40 ชุดมักทำให้เกิดโรคฮันติงตันที่มีเพเนแทรนซ์ไม่เป็น 100% และเริ่มมีอาการที่อายุมากกว่าชนิดอื่น ดำเนินโรคช้ากว่าชนิดอื่น ผู้ป่วยบางรายเริ่มมีอาการช้ามากจนไม่มีอาการอื่นๆ ปรากฏให้เห็นเลยก็มี ในขณะเดียวกันหากมีจำนวนชุด CAG ซ้ำเยอะมาก จะทำให้โรคฮันติงตันนั้นมีเพเนแทรนซ์ 100% และเริ่มแสดงอาการตั้งแต่อายุน้อยกว่า 20 ปี ซึ่งจะเรียกว่าโรคฮันติงตันชนิดวัยเด็ก หรือชนิด akinetic-rigid หรือชนิดเวสท์ฟาล ซึ่งคิดเป็นประมาณ 7% ของพาหะโรคฮันติงตันทั้งหมด
การถ่ายทอด
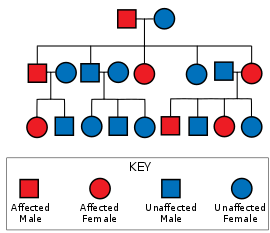
โรคฮันติงตันมีการถ่ายทอดแบบออโตโซมลักษณะเด่น หมายความว่าผู้ที่ได้รับอัลลีลของยีนฮันติงตินซึ่งผิดปกติ (mutant allele) มาเพียงยีนเดียวจากสองยีนก็สามารถแสดงอาการของโรคได้ และโอกาสที่ผู้ที่มียีนผิดปกติจะแสดงอาการของโรคก็มีสูงเกือบ 100% เนื่องจากโรคฮันติงตันเป็นโรคที่มีเพเนแทรนซ์สูงมาก การถ่ายทอดแบบออโตโซมลักษณะเด่นนี้จะทำให้ทายาทของผู้ป่วยมีโอกาสได้รับอัลลีลที่ผิดปกติจากผู้ป่วยไป 50% ซึ่งโอกาส 50% นี้ ไม่เกี่ยวข้องกับเพศ
ลำดับซ้ำชุดสามของ CAG ที่ยาวเกินกว่า 28 ชุดจะมีความไม่เสถียรระหว่างการทำซ้ำของสายดีเอ็นเอ โดยยิ่งมีจำนวนชุดซ้ำมากก็ยิ่งมีความไม่เสถียรมาก ความไม่เสถียรนี้มักทำให้เกิดการเพิ่มขึ้นของจำนวนชุดซ้ำจากรุ่นสู่รุ่น (dynamic mutation) แทนที่จะมีการถ่ายทอดจำนวนชุดซ้ำโดยไม่มีการเปลี่ยนแปลง ลักษณะเช่นนี้ทำให้คนรุ่นถัดไปอาจมีจำนวนชุด CAG ซ้ำไม่เท่ากับรุ่นพ่อแม่ ดังนั้นพ่อแม่ที่ไม่เป็นโรคแต่มีจำนวนชุดซ้ำอยู่ในระดับกลาง (28-35 ชุด) หรือเป็นโรคแต่อยู่ในระดับที่มีเพเนแทรนซ์ไม่ถึง 100% (36-40 ชุด) อาจมีทายาทที่มีจำนวนชุด CAG ซ้ำมากกว่าที่รุ่นพ่อแม่มี จนได้รับการถ่ายทอดโรคฮันติงตันที่มีเพเนแทรนซ์เป็น 100% ได้ การมีจำนวนชุด CAG ซ้ำเพิ่มขึ้นจากรุ่นสู่รุ่นเช่นนี้ทำให้โรคอาจมีความรุนแรงมากขึ้น (เช่น เริ่มมีอาการเร็วขึ้น อาการดำเนินไปรวดเร็วมากขึ้น) ปรากฏการณ์เช่นนี้เรียกว่า genetic anticipation โดยความไม่เสถียรของลำดับ CAG นี้จะพบในกระบวนการสร้างอสุจิมากกว่ากระบวนการสร้างไข่ ดังนั้นอัลลีลที่ได้รับการถ่ายทอดมาจากแม่จะมีความยาวใกล้เคียงกันมากกว่าอัลลีลที่ได้รับการถ่ายทอดมาจากพ่อ ซึ่งมีโอกาสที่จะมีการเพิ่มขึ้นของจำนวนชุด CAG ซ้ำมากกว่า ส่วนการเกิดการกลายพันธุ์จนเป็นโรคฮันติงตันซึ่งเกิดขึ้นใหม่ในคนที่พ่อแม่มีลำดับ CAG ซ้ำไม่เกิน 36 ชุดนั้นพบได้น้อยมาก
ผู้ป่วยที่มียีนผิดปกติทั้งสองอัลลีลนั้นมีจำนวนน้อยมาก โดยอาจพบได้ในครอบครัวขนาดใหญ่ที่มีการแต่งงานภายในเครือญาติ ก่อนหน้านี้เชื่อกันว่าโรคฮันติงตันเป็นโรคเพียงโรคเดียวที่การมีจำนวนยีนผิดปกติเป็นสองอัลลีลนั้นไม่ส่งผลต่ออาการและการดำเนินโรค แต่ปัจจุบันนี้พบแล้วว่าการมียีนผิดปกติสองอัลลีลนั้นส่งผลต่อลักษณะปรากฏหรือฟีโนไทป์และความเร็วของการดำเนินโรคด้วย ทายาทของผู้ป่วยที่มียีนผิดปกติทั้งสองอัลลีลนั้นจะต้องได้รับยีนผิดปกติมาอันหนึ่งอย่างแน่นอน ดังนั้นจะมีโรคฮันติงตันอย่างแน่นอน ในขณะที่ทายาทของพ่อแม่ที่เป็นโรคฮันติงตันแบบมียีนผิดปกติหนึ่งอัลลีลทั้งคู่นั้นมีโอกาสเป็นโรค 75% และมีโอกาสไม่เป็นโรค 25% โดยมีโอกาสเป็นโรคฮันติงตันแบบมียีนผิดปกติทั้งสองอีลลีลอยู่ 25% โดยทั่วไปแล้วแฝดเหมือนที่ได้รับยีนผิดปกติที่เหมือนกันมานั้น จะเริ่มแสดงอาการและมีการดำเนินโรคไม่เหมือนกัน
การวินิจฉัย
การวินิจฉัยแยกโรค
90% ของการวินิจฉัยโรคฮันติงตันขึ้นกับอาการและประวัติครอบครัวซึ่งมีลักษณะเฉพาะ ยืนยันได้ด้วยการตรวจทางพันธุศาสตร์พบมีการขยายของลำดับนิวคลีโอไทด์ที่ซ้ำกันซึ่งเป็นสาเหตุของโรค ยังมีโรคอื่นๆ ที่เรียกว่าโรคซึ่งคล้ายโรคฮันติงตัน (HD-like disorder) โรคเหล่านี้มักถูกเรียกรวมๆ ว่า HDL หรือ HD-like ("คล้ายโรคฮันติงตัน") สาเหตุของ HDL ส่วนใหญ่นั้นยังไม่เป็นที่ทราบ บางโรคที่ทราบสาเหตุนั้นพบว่าเกิดจากการกลายพันธุ์ของยีนพรีออนโปรตีน (HDL1) ยีนจังท์โตฟิลลิน 3 (HDL2) ยีน HTT ชนิดที่ถ่ายทอดแบบลักษณะด้อย (HDL3 พบในรายงานครอบครัวผู้ป่วยเพียงครอบครัวเดียว และยังไม่เป็นที่เข้าใจมากนัก) และยีนที่ถอดรหัสเป็น TATA box-binding protein (HDL4/SCA17) โรคที่ถ่ายทอดทางออโตโซมลักษณะเด่นอื่นๆ ที่อาจถูกวินิจฉัยผิดไปเป็นโรคฮันติงตันได้เช่น dentatorubral-pallidoluysian atrophy (การฝ่อของเดนเตโตรูบราล-พาลลิโดลุยเซียน) และ neuroferritinopathy (ความผิดปกติของเฟอร์ไรตินในระบบประสาท) นอกจากนี้ยังมีโรคที่ถ่ายทอดทางออโตโซมลักษณะด้อยอีกจำนวนหนึ่งที่อาจแสดงอาการคล้ายผู้ป่วยฮันติงตันที่ไม่ได้เกิดจากการถ่ายทอดในครอบครัว (sporadic case) เช่น chorea acanthocytosis (เม็ดเลือดแดงอะแคนโธซัยต์มากผิดปกติร่วมกับมีโคเรีย), pantothenate kinase-associated neurodegenartion (การเสื่อมของประสาทซึ่งสัมพันธ์กับแพนโทธีเนท ไคเนส) และ X-linked McLeod syndrome (กลุ่มอาการแม็คลีอ็อดซึ่งสัมพันธ์กับโครโมโซมเอกซ์)
การวินิจฉัยทางคลินิก

การตรวจร่างกายและการตรวจสภาพจิตสามารถช่วยบอกได้ว่าผู้ป่วยเริ่มมีอาการของโรคแล้วหรือยังไม่มี อาการที่มักนำผู้ป่วยมาพบแพทย์มากที่สุดคือการเคลื่อนไหวผิดปกติของส่วนต่างๆ ของร่างกาย หากอาการเหล่านี้เป็นขึ้นทันทีทันใด เริ่มเป็นแบบไม่สัมพันธ์กับเหตุการณ์หรืออาการอื่นๆ จะเป็นการชี้ให้ควรสงสัยว่าต้องให้การวินิจฉัยผู้ป่วยเป็นโรคฮันติงตัน อาการทางการรับรู้หรือทางสภาพจิตมักไม่เป็นอาการแรกๆ ที่ทำให้นึกถึงโรคฮันติงตัน ส่วนใหญ่จะเป็นจะนึกถึงอาการเหล่านี้ว่าเป็นจากโรคฮันติงตันก็ต่อเมื่อเป็นการได้ประวัติย้อนหลังของผู้ป่วยโรคฮันติงตันที่วินิจฉัยได้จากสาเหตุอื่น หรืออาการเหล่านี้ปรากฏขึ้นหลังจากได้รับการวินิจฉัยเรียบร้อยแล้ว แพทย์สามารถวัดการดำเนินโรคออกมาเป็นตัวเลขได้โดยใช้ unified Huntington's disease rating scale (เกณฑ์การให้คะแนนโรคฮันติงตันแบบสากล) โดยคะแนนนี้คิดจากการประเมินผู้ป่วยทั่งด้านการสั่งการ พฤติกรรม การรับรู้ และความสามารถในการดำเนินชีวิตการถ่ายภาพรังสีทางการแพทย์เช่นซีทีสแกนหรือเอ็มอาร์ไออาจแสดงให้เห็นได้เพียงภาวะสมองใหญ่ฝ่อที่จะพบก็ต่อเมื่อโรคดำเนินไปมากแล้วเท่านั้น การถ่ายภาพรังสีของการทำงานของระบบประสาท เช่น fMRI หรือ PET scan อาจแสดงให้เห็นการเปลี่ยนแปลงของการทำงานของสมองก่อนที่จะปรากฏอาการได้ แต่วิธีนี้ยังมีที่ใช้เฉพาะในการศึกษาวิจัย ยังไม่มีการนำมาใช้ทางคลินิก